보도자료 홈
산업별
주제별
지역별
상장사
사진
뉴스와이어 제공
이 보도자료는 게재 기간이 끝나 뉴스와이어에서 볼 수 있습니다.
뉴스와이어에서 보도자료 보기 >
인기 토픽
신제품 출시
휴가
인공지능
바이오테크
스타트업
상장사 보도자료
GS샵, 폭염에 ‘과채주스’ 주문액 260% 증가… 과일·채소 대신 간편하게
대웅제약, 스타강사 이지영과 손잡고 ‘임팩타임 A+ 30포’ 한정판 출시… 수능 D-100 수험생 맞춤 응원 나선다
삼성전자, 8세대 갤럭시 폴더블 기술 혁신 담은 ‘폴더블 헤리티지’ 전시
NHN KCP, 2분기 역대 최대 매출 3834억·거래액 17조 달성… 영업이익 154억
팀뷰어, 자율형 워크플레이스 조사 발표… 신뢰 격차 해소가 AI 생산성 향상의 관건
전시회
2026 부산국제사진제
2026 부산 세계도서관정보대회
2026 부산국제주류박람회
2026 제5회 제주비엔날레
2026 부산국제마케팅광고제(MAD STARS 2026)
동영상
서울광역새일센터, ‘2026 멈추지 않는 커리어 숏폼 공모전’ 시상식 개최
인기 사진
이전
다음
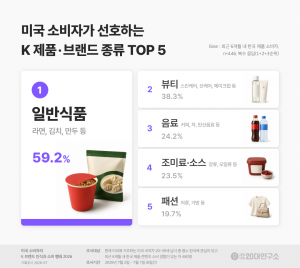
미국 소비자가 선호하는 K 제품·브랜드 종류 TOP 5